

This friday it’s World Sickle Cell Awareness Day. This debilitating disease affects millions of people world wide. Luckily, scientists have a sound understanding of the genetic cause of Sickle Cell Disease (SCD) which makes the disease easier to trace, treat, and – with continued research – cure.

SCD is caused by small changes in an individual’s genetic coding that interfere with their body’s ability to create hemoglobin – a protein that transports oxygen. While most diseases and health conditions have some type of genetic component as well as environmental components, SCD is caused by mutations to a single gene in all the cells of a person’s body. Such diseases are called monogenic. Because SCD can be traced back to a single gene, doctors are able to identify, trace, and treat this disease using biotechnologies like PCR and CRISPR.
However, SCD still affects millions of people throughout the world. It is particularly prevalent in Sub-Saharan Africa, South America, Central America, Saudi Arabia, India, and the Mediterranean. SCD is also more commonly found in people whose ancestors come from these places. This may be because inheriting one mutated sickle cell gene actually helps protect people from another serious disease – malaria – while causing few SCD symptoms.
What are the symptoms of SCD?
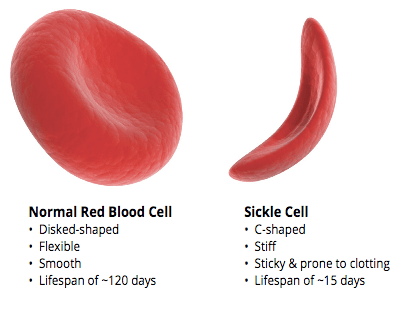
The symptoms of SCD usually appears 5-6 months after birth. In children, the disorder can cause delayed growth and development, yellowing of the skin and eyes, secondary infections like pneumonia and episodes of pain called “pain crisis”. If left untreated these symptoms worsen with time.
Adults with severe SCD can experience chronic pain, fatigue, aseptic necrosis (localized bone death), and vision loss; as well as life-threatening lung and heart injuries. In less severe cases, the main symptoms are tiredness, dizziness, difficulty breathing, and a weakened immune system.
The source of all these symptoms are modified/misshaped hemoglobin proteins that alter a person’s red blood cells. Individuals with SCD have red blood cells that are misshaped, stiff, sticky, and short-lived.
What causes SCD?
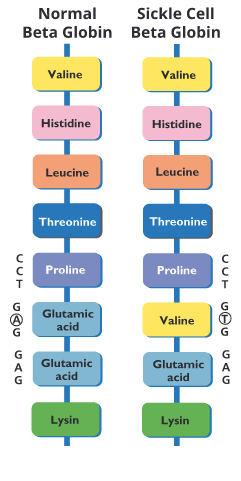
Researchers have identified around 400 different DNA changes that can lead to SCD. The most common is an A to T single nucleotide mutation that alters the amino acid sequence of a protein known as beta-globin. All SCD mutations occur in this gene.
Recessive diseases like SCD can “skip” multiple generations. This happens whenever there is a generation of carriers – heterozygous individuals with a dominant normal gene that masks the continued presence of the disease-causing mutation. However, such individuals can have children that do have the disease. The possibility of a child having this disease can be predicted by using a two-by-two grid known as a Punnett Square. Sometimes couples who are considering starting a family will go to a genetic counselor in order to determine their genotypes at the beta-goblin gene and the possible genotypes of any children.
The beta-globin gene is found on chromosome 11 and contains the body’s only set of instructions for making an essential part of most hemoglobin proteins. Because it is located on an autosomal (or paired) chromosome, each individual has two copies – one that they inherited from their mother and one that they inherited from their father. Humans need only one “normal” beta-goblin gene to produce a sufficient supply of healthy hemoglobin. In such a case the normal gene is called “dominant” because its effects can compensate for a mutated gene. Similarly, SCD is called a “recessive” disease because symptoms only appear when someone has inherited two mutant versions of the beta-globin gene.
How is SCD being treated?
Early and accurate identification of SCD is the first step in treating SCD. Many places in the US now automatically screen newborns for sickle cell disease either by measuring the level of hemoglobin in a blood sample, observing red blood cells under a microscope, or sequencing a DNA sample.
Once identified sickle cell symptoms can be treated with a combination of medicines such as Hydroxyurea and antibiotics, medical treatments blood cell transfusions, and lifestyle changes. These not only help individuals address their symptoms but also minimize the long term effects that repeated episodes of low oxygen can have on their organs.
Medical researchers are also currently working to find a cure for this disease. One current option is a bone marrow transplant. However, this requires a closely matched marrow donor, several rounds of chemotherapy, and years of taking immunosuppressive drugs. Consequently, transplants are saved for severe cases of SCD in otherwise healthy and young patients. Another potential cure is gene therapy – using biotechnologies like CRISPR to introduce a healthy copy of the mutated hemoglobin gene into a patient’s genome.
In the summer of 2019 Victoria Gray underwent an experimental treatment called CRISPR where researchers collected some of her bone marrow cells, edited their genetic material to produce fetal hemoglobin, destroyed most of her unedited bone marrow cells, and then reintroduced the edited cells. The initial results were promising – Ms. Gray observed a dramatic reduction in most of here SCD symptoms and early blood tests showed healthy hemoglobin and red blood cell levels.
A major goal of this day is to increase public knowledge and understanding of this disease.
Interested in supporting this goal? Visit the Sickle Cell Disease Association of America’s Shines the Light on Sickle Cell webpage. Or consider incorporating one of these experiments into your classroom: Sickle Cell Gene Detection, In Search of the Sickle Cell Gene by Southern Blot, or our soon to be released Bioinformatics and Python exercise.

3 comments
Comments are closed.